Research
Natural products chemistry: Total syntheses, biological activities, and analysis of structure-activity relationships
Secondary metabolites from natural sources have manifold biological activities in living organisms, and have undergone optimization by nature over thousands of years. Hence, due to their bioactivity, but also their structural diversity natural products represent a most attractive source of lead structures for drug development.
We work out total syntheses of natural products with cytotoxic and/or antimicrobial as well as cation channel-modulatory activities (e.g. β-carbolines, 4-quinolones, benzylisoquinolines, polycyclic aromatic alkaloids, steroid alkaloids) from tropical plants (e.g. Annonaceae) and from marine organisms (tunicates, sea anemones, sponges). Hereby, we focus on natural products with drug-like structures, i.e. manageable structural complexity and promising physicochemical properties (following Lipinski’s rule of five). Compounds with significant biological activities are being further modified in order to optimize their activity and selectivity, and to gain deeper insight into structure-activity relationships.
A number of these natural products served as lead structures for the development of bioactive compounds for epigenetic targets (kinase inhibitors, bromodomain inhibitors, histone deacetylase inhibitors, …; see below) in our recent work.
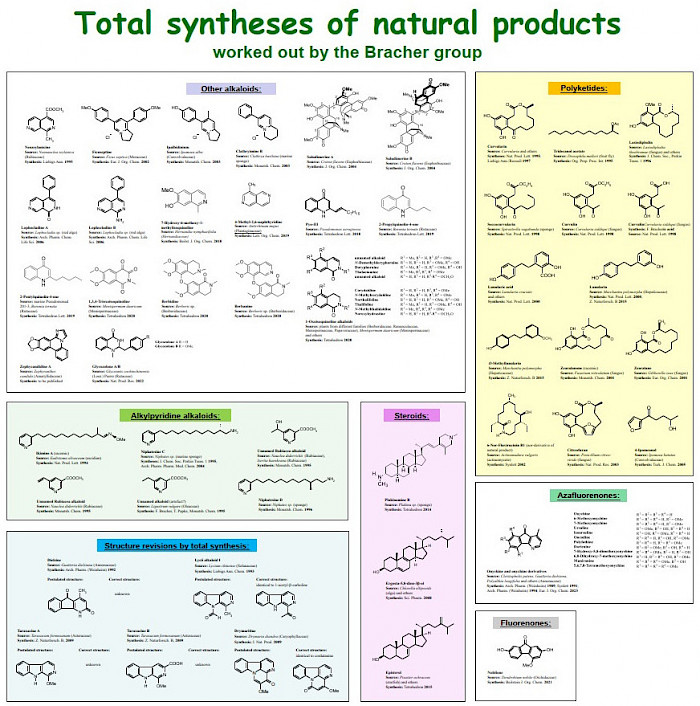
For a compilation of all natural products total syntheses of the Bracher group over three decades, see extra file Total Syntheses Bracher
An overview of all total syntheses of natural products performed by the Bracher group is shown below.
Click on image to enlarge.

Epigenetic targets: Kinases, lysine deacetylases, and others
The regulation of transcription of genetically encoded information is essential for all living organisms. Numerous posttranslational modifications of DNA and proteins (e.g. histones, tubulin) are part of this regulation. These processes open new and attractive targets for new drugs for treating different diseases (cancer, inflammatory diseases, infections).
In cooperation with diverse partners we develop new bioactive compounds for epigenetic targets. In many cases, natural products from our own research (see above) acted as lead structures for these investigations.
Our most important targets are:
Kinases:
A number of natural products for which total syntheses had been developed in our group were found to be inhibitors of kinases. Starting from the β-carboline alkaloids bauerine C and annomontine we developed potent and selective inhibitors of the kinases CLK1, CLK3, PIM1, DYRK1A, and BMP2K.
Further characterization of these inhibitors was/is being performed in cooperation with the following groups: Prof. Dr. S. Knapp (Structural Genomics Consortium, Oxford, now Frankfurt/Main), Prof. Dr. P. Filippakopoulos (Oxford), Prof. Dr. J. Schwaller (Basel), Dr. L. Meijer (ManRos, Roscoff), Prof. Dr. W. Becker (Aachen), Prof. Dr. T. Meyer (MPI für Infektionsbiologie, Berlin), Prof. Dr. O. Livnah (Jerusalem), Prof. Dr. G. Pradel (Aachen), Prof. Dr. A. Vollmar (LMU), Prof. Dr. S. Zahler (LMU), Prof. Dr. A. Aigner (Leipzig), Prof. Dr. U. Rauch-Kroehnert (Berlin), Prof. Dr. Robert Fürst (Frankfurt/Main), Prof. Dr. Wolfgang Link (Madrid), and others.
Below you find structures of selected kinase inhibitors from our projects:
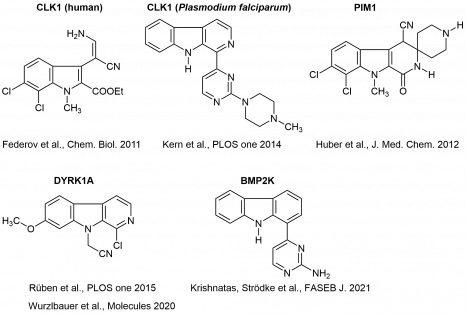
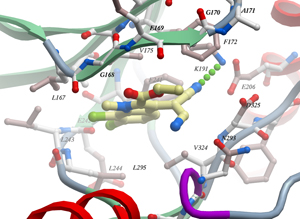
Co-crystallizations of inhibitors with kinases are performed in cooperation with Stefan Knapp at the Structural Genomics Consortium (Oxford, UK), now Goethe-Universität Frankfurt, and in the group of Oded Livnah (Jerusalem, Israel). Data are published in the Protein Data Bank (PDB) (see „Publications“).
An outstanding result of these investigations was the discovery of a novel, uncommon binding mode of chlorinated indole-type inhibitors to the hinge region of kinases by halogen bonding (Huber et al., 2012).
Lysine deacylases (KDACs, HDACs):
The interaction of DNA and histones is controlled on an epigenetic level by modifications of histone proteins (methylation, acetylation, …). The extent of lysine acetylation is a major factor determining gene regulation. Inhibitors of histone deacetylases open new opportunities in tumor therapy, but also in fighting microbial and protozoal infections.
In cooperation with Prof. Dr. M. Jung (Freiburg), Prof. Dr. W. Sippl (Halle), Prof. Dr. C. Steegborn (Bayreuth) and groups in the SFB 1309 we develop novel inhibitors of histone deacetylases (sirtuins, HDAC6).
In cooperation with Prof. W. de Souza (Rio de Janeiro) and Prof. Dr. R. Pierce (Lille) we could demonstrate the potential of sirtuin inhibitors for treatment of protozoal infections.
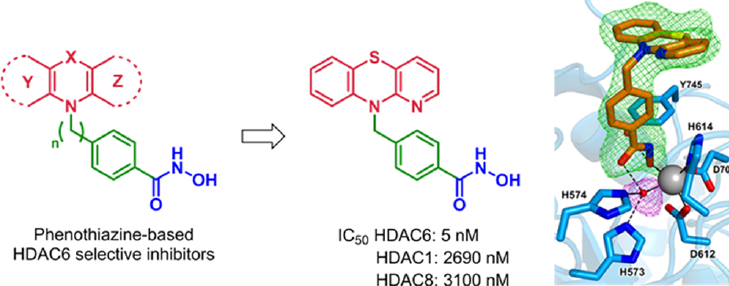
Phenothiazine-based HDAC6 inhibitors (Vögerl et al., J. Med. Chem. 2019)
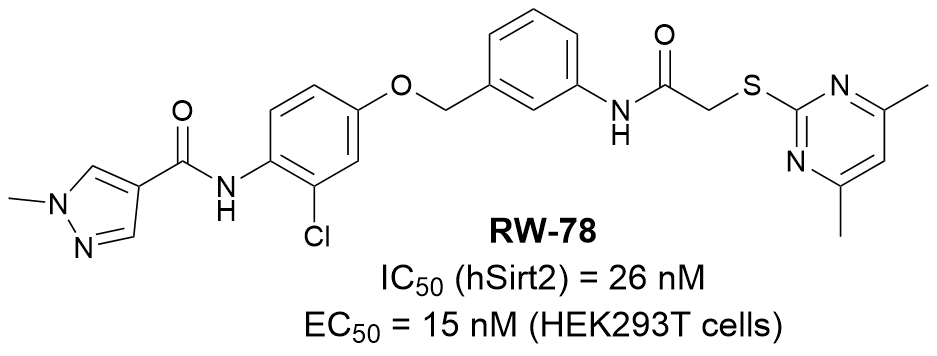
Sirtuin 2 is one of the seven sirtuin subtypes that emerged as a key target due to its broad catalytic scope and diverse substrates and thus its involvement in the pathogenesis of various diseases such as neurodegeneration, cancer and viral infections. Based on the lead compound SirReal2 and the advanced SirReal-type compound 24a, we developed a series of highly potent and subtype selective sirtuin 2 inhibitors through various drug design strategies such as molecular rigidization (Frei et al. Molecules. 2025), molecular hybridization (Frei et al. Int. J. Mol. Sci. 2025) and rational SAR functional group modifications (Wirawan et al. RSC. Med. Chem. 2025).
In cooperation with Prof. Dr. Michael Groll and PD Dr. Eva Huber (TUM) within SFB1309, we demonstrated through X-ray co-crystallography a novel binding mode of the highly potent and subtype selective sirtuin 2 inhibitor RW-78 (in green) that forms halogen-π interactions with sirtuin 2 residues and interferes with NAD+ co-factor binding. In cooperation with Prof. Dr. Manfred Jung (Freiburg), we also demonstrated high target engagement of RW-78 on a cellular level.
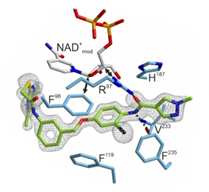
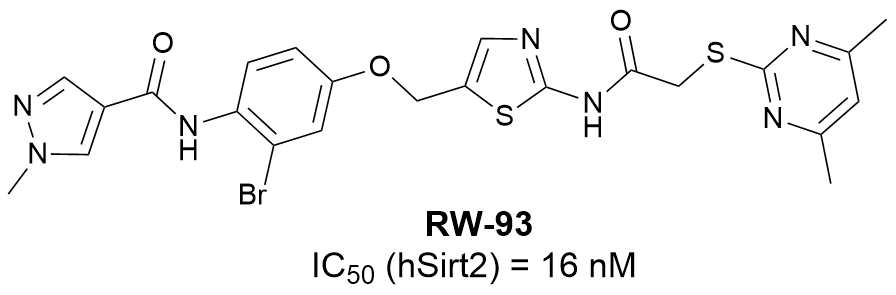
Further efforts focusing on the optimization of sirtuin 2 inhibitors through molecular hybridization led to the development of the highly potent and subtype selective RW-93 with an IC50 (hSirt2) of 16 nM, making RW-93 the most potent low-molecular sirtuin 2 inhibitor known to date.
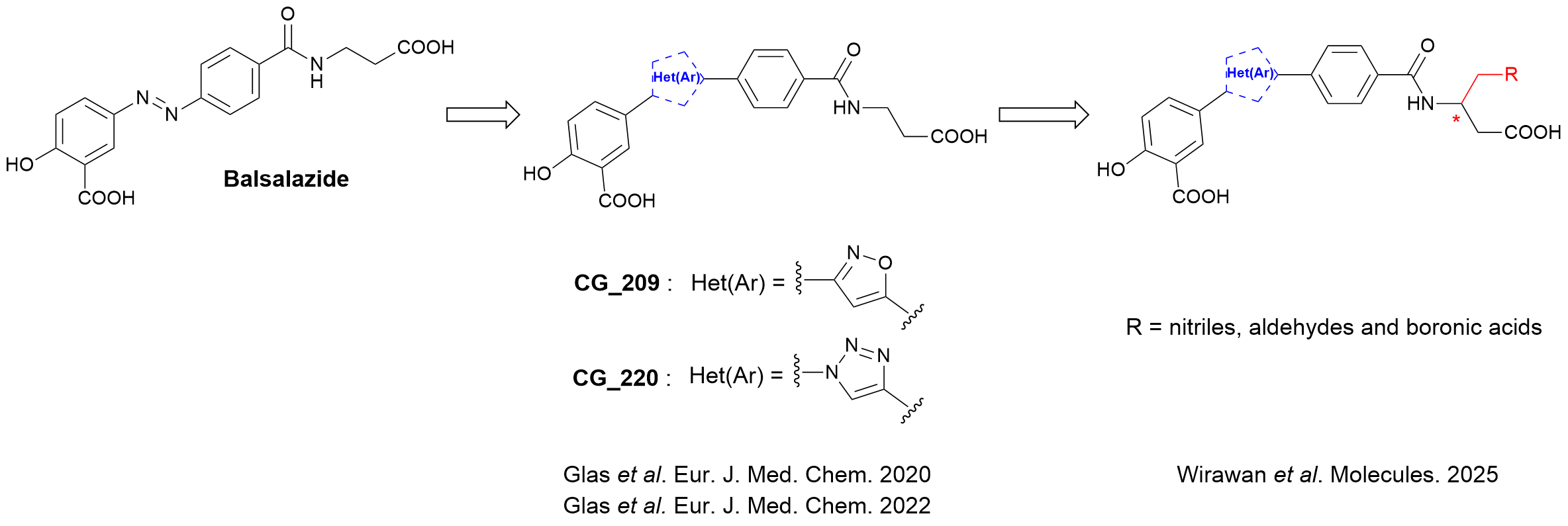
Sirtuin 5 is a very special, hitherto rather underexplored NAD+-dependent lysine deacylase, which preferentially removes acyl residues derived from dicarboxylic acids from lysine residues. Based on the anti-inflammatory drug balsalazide, a hit from a HTS, we developed novel, selective inhibitors of this sirtuin subtype as valuable chemical tools that incorporate the metabolically more stable heteroaromatic rings in place of the labile azo bond (Glas et al. Eur. J. Med. Chem. 2020, Glas et al. Eur J. Med. Chem. 2022).
Based on this optimized chemotype, we systematically investigated enantiomerically pure derivatives bearing electrophilic functional groups such as boronic acids, nitriles and aldehydes that were rationally positioned on the inhibitor scaffold via molecular docking experiments to potentially enhance their potency through reversible covalent bonding with the co-factor NAD+ (Wirawan et al. Molecules 2025).
Bromodomains:
Bromodomains are epigenetic „readers“ which are involved in the regulation of N-acetylation of lysine residues in histones, and consequently in gene regulation. Bromodomain inhibitors represent potential drugs for treatment of different tumors, but also other diseases.
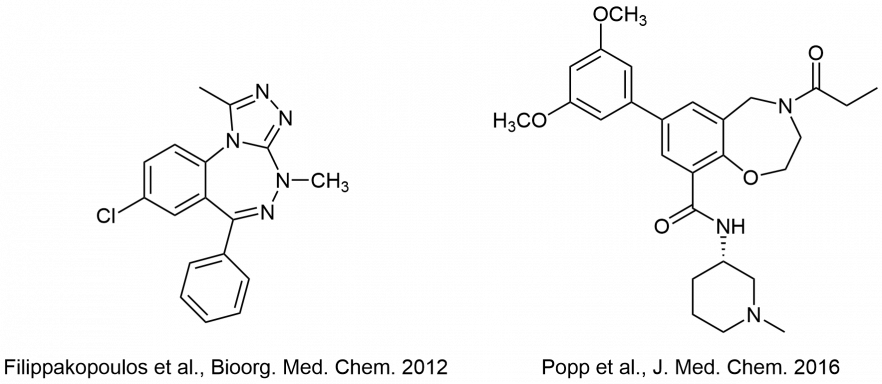
In cooperation with the Structural Genomics Consortium (Oxford, UK) we developed inhibitors of several types of bromodomains (BRD, CREBBP, …).
Some bromodomain inhibitors from the Bracher lab:
Macrodomains:
Macrodomains are involved in the regulation of ADP-ribosylation of proteins and in gene transcription.
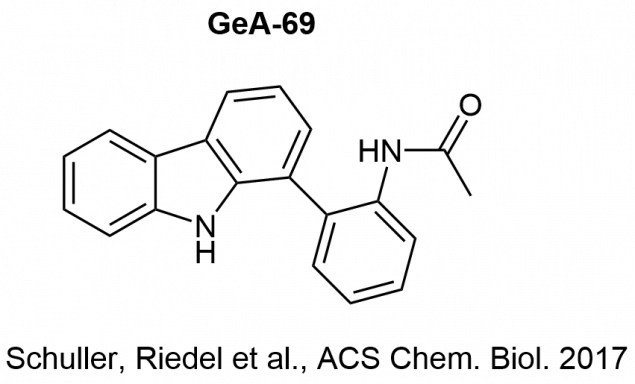
In cooperation with the Structural Genomics Consortium (Oxford, UK) we developed arylcarbazoles (GeA-69) as a new chemotype of macrodomain PARP14 inhibitors.
Steroidomics
(together with Dr. Christoph Müller)
Synthesis and analysis of steroids, development of sterol biosynthesis inhibitors
Inhibitors of ergosterol biosynthesis as new antifungals
Substances inhibiting enzymes in the biosynthesis of ergosterol, the main sterol in membranes of fungi, represent the most important class of antifungals. Until now only four of these enzymes are used as drug targets for antifungals. Our research is mainly aimed at the imitation of cationic “high energy intermediates“ of crucial steps of ergosterol biosynthesis, what results in inhibition of selected enzymes (e.g., Δ14-reductase, Δ8,Δ7-isomerase, C24-sterol methyltransferase, oxidosqualene cyclase, C22-desaturase, ...).
We worked out a cell-based assay for the identification of target enzymes in ergosterol biosynthesis in fungi, based on incubation of model strains with the potential enzyme inhibitors, followed by cell lysis and analysis of the modifications in the sterol patterns by GLC-MS/MS. For quantitation of the inhibitory activity we developed a GC-MS/MS protocol which does not afford the use of radioactive material (Müller et al., Nature Protocols 2017).
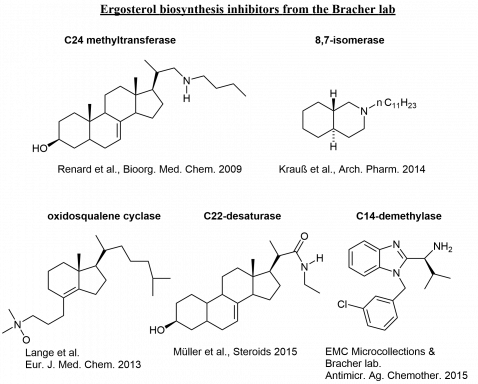
A collection of ergosterol biosynthesis inhibitors from our projects:
Inhibitors of cholesterol biosynthesis as new antilipidemic drugs – and beyond!
In analogy to the screening system described above, we worked out a whole cell assay for the identification of inhibitors of cholesterol biosynthesis in human cells. For the quantitation of the inhibitory effect we once again use 13C-isotope labeling and GC-MS/MS analysis (Müller et al., Nature Protocols 2019).
In the course of this research the first selective inhibitor of the enzyme lathosterol oxidase was developed. So the last gap concerning inhibitors of enzymes of the post-squalene part in cholesterol biosynthesis was closed.
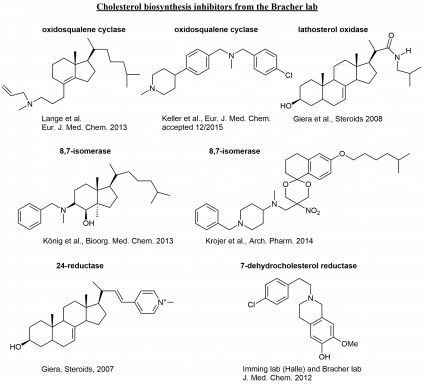
A collection of cholesterol biosynthesis inhibitors from our projects:
For these assays we need intermediates of the biosynthesis pathways as reference standards. If necessary, new synthesis approaches to these sterols are worked out in our lab (Giera & Bracher 2008, Dittrich & Bracher 2015).
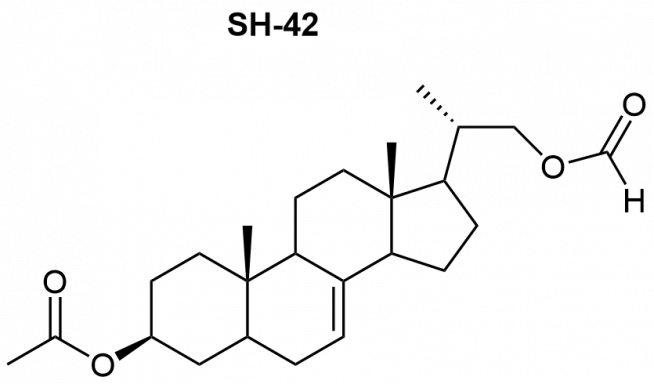
Recently, we identified inhibitors of the enzyme Δ24-dehydrocholesterol reductase (DHCR24) like SH-42 as novel anti-inflammatory compounds with a completely new molecular mode of action (Müller et al., Eur. J. Med. Chem. 2017; Körner et al., PNAS 2019: review article: Müller et al., Curr. Med. Chem. 2022).
Analysis of steroids
Based on the expertise we gained in the installation of the sterol biosynthesis assays, we have outstanding experience in the analysis of steroids by means of high-end GC-MS/MS, including modern sample preparation techniques from complex matrices (SPE, QuEChERS, SPME). This expertise is highly appreciated by numerous cooperating groups for whom we identify and analyze steroids (ergosterol, cholesterol and metabolites, cholic acids, phytosterols) in cells, cell cultures, blood, tissues, feces, plant material, extracts, and drug preparations (e.g., Müller at al., Planta Med. 2015; Müller at al., Molecules 2018; Junker et al., Molecules 2019).
Our analytical experience is also applied (guided by Dr. Christoph Müller) to some cooperative projects for ultra trace analysis of drugs, toxins (e.g. nicotine), and pesticides in food, surface water, and environmental samples (Müller et al., J. Food Sci. 2014; Vetter et al., 2017; Stöckelhuber et al., 2017; Löbbert et al., Analytical Methods 2021; Schanzer et al., Analytical Methods 2021).
Modulators of calcium channels
In cooperation with Prof. Dr. Christian Grimm (LMU, Department of Medicine), Prof. Dr. Martin Biel (LMU, Department of Pharmacy), and Prof. Dr. Angelika Vollmar/Dr. Karin Bartel (LMU, Department of Pharmacy), we develop novel inhibitors of lysosomal calcium channels (TPCs, TRPMLs) as valuable chemical tools for the in-depth characterization of the ion channels, but also as potential lead structures for the development of drugs.
In this effort, both activators and inhibitors are of interest, and our synthetic chemistry is predominantly aimed at improvement of activity and selectivity of modulators, which were detected in high-throughput screenings of large substance libraries.
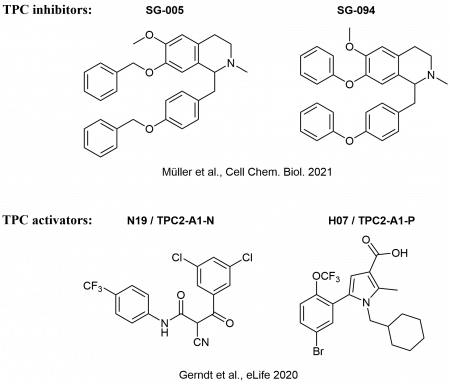
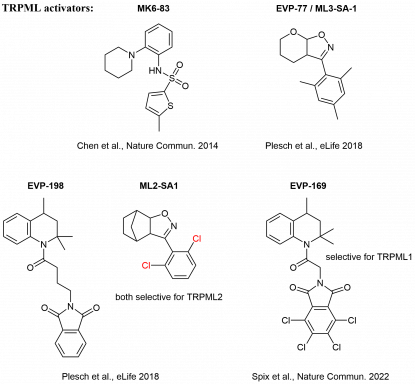
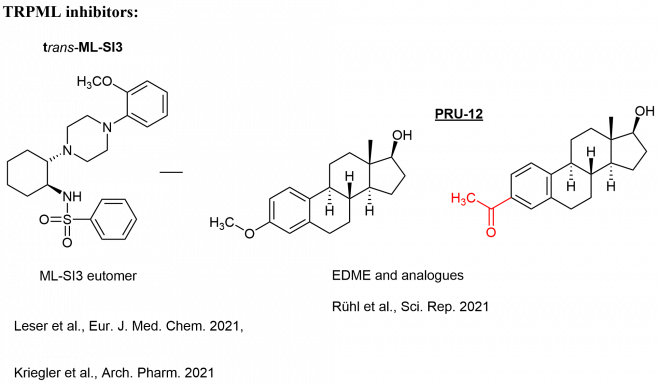
The new modulators are investigated as potential drugs for treatment of genetic diseases (like Mucolipidosis Type IV) and cancer (Chen et al., Nature Commun. 2014; Plesch et al., eLife 2018, Müller et al., Cell Chem. Biol. 2021).
Selected ion channel modulators from the Bracher lab: